Since the discovery of sickle-shaped cells in human blood, there have been a few major milestones in the treatment and efforts to bring awareness to the disease. Below is a historical timeline of these major milestones.
THE HISTORY OF SICKLE CELL
First Description of Sickle-Shaped Blood Cells by Dr. James Herrick
In 1904, Walter Clement Noel travelled from Grenada to the United States to start studying at the Chicago College of Dental Surgery. A few months later he was admitted to the Presbyterian Hospital in Chicago when he developed severe respiratory distress and a leg ulcer, both of which we now..Read MoreGenetic basis for SCD observed by Dr. V. Emmel
The third case of Sickle Cell was described in 1915 by Cook and Meyer in a 21-year-old woman. Interestingly, blood samples from both the patient and her father, who displayed no symptoms, showed the sickling deformity of the red cells and three of her siblings had died from severe anaemia. These..Read MoreDr. V.R Mason names the disease Sickle Cell Anemia
Dr. Mason, who observed the fourth reported case of Sickle Cell, was also the first to call the disease “Sickle Cell Anemia” and to notice the similarities between the cases. He also noted that all of these patients were black, inadvertently giving rise to the popular misconception that Sickle Cell originated..Read MorePathophysiology of Sickling Explained
Hahn and Gillespie were the first to associate the red cell sickling to low oxygen and acidic conditions. They were able to revert sickled cells back to their normal discoid shape by simply providing the cells with oxygen. Further experiments showed that, apart from oxygen, increased serum acidity also induced..Read MoreThe Protective Role of Fetal Haemoglobin
The protective role of fetal haemoglobin (HbF) was discovered in the 1940s, when Dr. Janet Watson suggested a link between HbF levels and the presence of disease symptoms in 1948. She observed that higher HbF levels in newborns kept them asymptomatic. Humans possess predominantly HbF in their fetal life but..Read MoreSickle Cell Becomes the First Molecular Disease Discovered
Work from several scientists contributed to the discovery of Haemoglobin, the protein responsible for Sickle cell disease. In 1940 Irwin Sharman noticed a difference between the way light passed through sickled blood cells compared to normal cells. Dr. Castle, a Harvard professor in Medicine, understood the implication of this finding:..Read MoreSickle Cell Trait and Protection from Malaria
Many of the studies that examined the association between the sickle cell trait and malaria stemmed from the question, “Why is the sickle cell trait maintained in such high frequency when the homozygous mutations (two genes with Sickle mutation) result in death?” One possible explanation was that the heterozygous trait..Read MoreSickle Hemoglobin Sequenced
Haemoglobin is an iron-containing protein that transports oxygen. There are two parts to this protein: 1) the heme component which consists of the iron and the globin component which consists of the globin protein. The heme groups were identical between HbA and HbS suggesting that the differences should be in..Read MoreSickle Gene Mapped
Development of DNA sequencing by Walter Gilbert and Frederick Sanger allowed the mapping of the sickle cell gene. DNA is made up of four different bases (these are the letters of the DNA alphabet A, T, C and G) and the genes coded in the DNA are identified (or read)..Read MorePenicillin Recognized as Preventative Medicine for Children with Sickle Cell
Loss of splenic function (see Disease complications and Treatment section for more information) makes Sickle cell patients more susceptible to bacterial infections, especially pneumococci which results in Pneumococcal meningitis. In the early 1980s, it was noted that these Pneumococcal infections can be prevented by prophylactic penicillin in early childhood and..Read MoreNational Sickle Cell Awareness Month Instituted in the USA
September is National Sickle Cell Awareness Month. The annual observance originated in 1975 when the National Association for Sickle Cell Disease (NASCD) and its member organizations began conducting month-long events to raise awareness about sickle cell disease and the need to address the problem at the national and local level. The..Read MoreFirst Reported Cure for Sickle Cell: Bone Marrow Transplantation
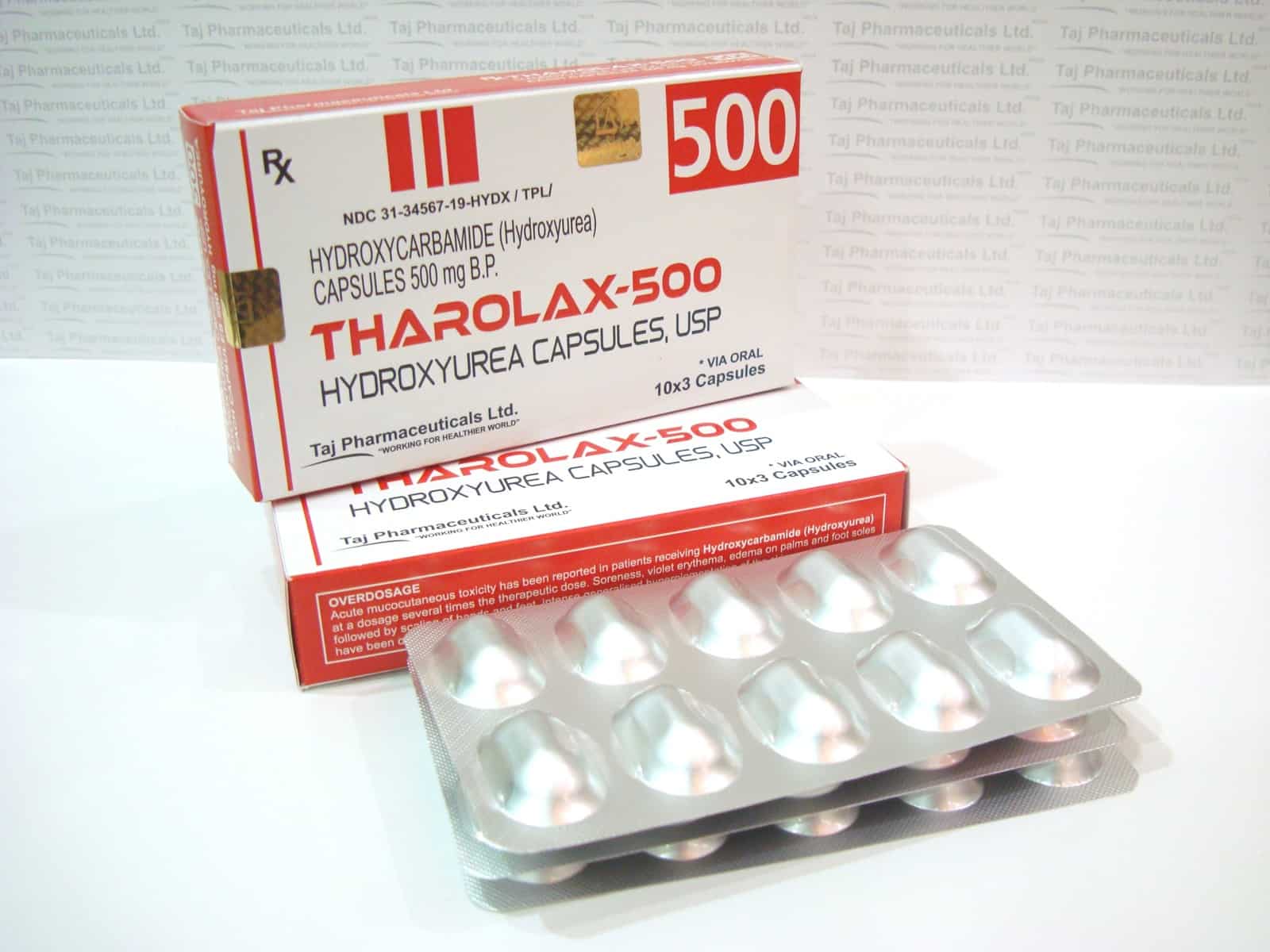
The first reported case of using a bone marrow transplantation to cure sickle cell disease was performed on an 8-year-old girl that also had acute leukemia. The bone marrow of the girl with SS was successfully replaced with bone marrow of her AS brother. Since then, bone marrow transplants have..Read MoreFDA Approves the Use of the Drug Hydroxyurea to Treat Sickle Cell
Much of the pharmacological research to treat sickle cell focused on drugs that would induce the production of fetal haemoglobin because of its protective role. The first drug to be tested in patients that showed an increase in fetal haemoglobin production was 5-azacytidine, but this drug was never tested in..Read MoreSetback in Gene Therapy Jesse Gelsinger
Because sickle cell anaemia is caused by one defective gene, it is a good candidate for gene therapy. If the sickle cell gene can be replaced by a normal gene in the bone marrow, patients will be able to produce normal haemoglobin. Scientists have been exploring this option for several..Read MoreNewborn Screening for Sickle Cell begins in Ontario
In November 2006, Hemoglobinopathy (HbSS, HbSC, and Hbs/ß-thalassemia) screening in newborns began in the province of Ontario.World Sickle Cell Day Established by the United Nations
World Sickle Cell Day was established by the United Nations General Assembly in 2008 in order to increase the awareness about the Sickle Cell Disease and its cure among the common public. It was celebrated first time on 19th of June in 2009.100th Anniversary of the Identification of Sickle Cell by Dr. Herrick
National Sickle Cell Awareness Day Act Passed by the Canadian Government
On November 22, 2017, the Canadian House of Commons passed Bill S-211 to designate the nineteenth day of June in each and every year as “National Sickle Cell Awareness Day”. The Bill received Royal Assent on December 12, 2017, making it an official law. The Bill was sponsored by Senator Jane..Read More




References
1. Haller, J.O., W.E. Berdon, and H. Franke, Sickle cell anemia: the legacy of the patient (Walter Clement Noel), the interne (Ernest Irons), and the attending physician (James Herrick) and the facts of its discovery. Pediatr Radiol, 2001. 31(12): p. 889-90.
2. Steensma, D.P., R.A. Kyle, and M.A. Shampo, Walter Clement Noel-first patient described with sickle cell disease. Mayo Clin Proc, 2010. 85(10): p. e74-5.
3. Herrick, J.B., Peculiar elongated and sickle-shaped red blood corpuscles in a case of severe anemia. Archives of Internal Medicine, 1910. 6: p. 517–521.
4. Herrick, J.B., Peculiar elongated and sickle-shaped red blood corpuscles in a case of severe anemia. 1910. Yale J Biol Med, 2001. 74(3): p. 179-84.
5. Serjeant, G.R., The emerging understanding of sickle cell disease. Br J Haematol, 2001. 112(1): p. 3-18.
6. Cook, J., Meyer, J, Severe anemia with remarkable elongated and sickle-shaped red blood cells and chronic leg ulcer. Archives of Internal Medicine, 1915. 16: p. 644-651.
7. Emmel, V., A study of the erythrocytes in a case of severe anemia with elongated and sickle shaped red blood corpuscles. Archives of Internal Medicine, 1917. 20: p. 586.
8. Foy, H., A. Kondi, and W. Brass, Sickle-cell disease of Africans in Kenya. East Afr Med J, 1951. 28(1): p. 1-5.
9. Mason, V., Sickle Cell Anemia. The Journal of the American Medical Association, 1922. 79: p. 1318-1320.
10. Lehmann, H., Distribution of the sickle cell gene : A new light on the origin of the East Africans. Eugen Rev, 1954. 46(2): p. 101-21.
11. Kamel, K.a.A., A, Origin of the sickling gene. Nature, 1965. 205: p. 919.
12. Kan, Y.W. and A.M. Dozy, Polymorphism of DNA sequence adjacent to human beta-globin structural gene: relationship to sickle mutation. Proc Natl Acad Sci U S A, 1978. 75(11): p. 5631-5.
13. Pagnier, J., et al., Evidence for the multicentric origin of the sickle cell hemoglobin gene in Africa. Proc Natl Acad Sci U S A, 1984. 81(6): p. 1771-3.
14. Kutlar, A., et al., Hematological observations on Arabian SS patients with a homozygosity or heterozygosity for a beta S chromosome with haplotype #31. Hemoglobin, 1985. 9(6): p. 545-57.
15. Wainscoat, J.S., et al., A novel deletion in the beta-globin gene complex. Ann N Y Acad Sci, 1985. 445: p. 20-7.
16. Hahn, E., Gillespie, EB, Sickle cell anemia. Report of a case greatly improved by splenectomy. Experimental study of sickle cell formation. Archives of Internal Medicine, 1927. 39: p. 233-254.
17. Brugnara, C., L. de Franceschi, and S.L. Alper, Inhibition of Ca(2+)-dependent K+ transport and cell dehydration in sickle erythrocytes by clotrimazole and other imidazole derivatives. J Clin Invest, 1993. 92(1): p. 520-6.
18. Brugnara, C. and D.C. Tosteson, Inhibition of K transport by divalent cations in sickle erythrocytes. Blood, 1987. 70(6): p. 1810-5.
19. De Franceschi, L., et al., Oral magnesium supplements reduce erythrocyte dehydration in patients with sickle cell disease. J Clin Invest, 1997. 100(7): p. 1847-52.
20. Watson, J., The significance of the paucity of sickle cells in newborn Negro infants. Am J Med Sci, 1948. 215(4): p. 419-23.
21. Huehns, E.R., et al., Human Embryonic Hemoglobins. Cold Spring Harb Symp Quant Biol, 1964. 29: p. 327-31.
22. Seltzer, W.K., et al., Molecular genetic studies in black families with sickle cell anemia and unusually high levels of fetal hemoglobin. Hemoglobin, 1992. 16(5): p. 363-77.
23. Miller, B.A., et al., High fetal hemoglobin production in sickle cell anemia in the eastern province of Saudi Arabia is genetically determined. Blood, 1986. 67(5): p. 1404-10.
24. Sherman, I., The sickling phenomenon, with special reference to the differentiation of sickle cell anemia from the sickle cell trait. Johns Hopkins Hospital Bulletin, 1940. 67: p. 309-324.
25. Daland, G.A. and W.B. Castle, A simple and rapid method for demonstrating sickling of the red blood cells; the use of reducing agents. J Lab Clin Med, 1948. 33(9): p. 1082-8.
26. Pauling, L., H.A. Itano, and et al., Sickle cell anemia a molecular disease. Science, 1949. 110(2865): p. 543-8.
27. Pauling, L., H.A. Itano, and et al., Sickle cell anemia, a molecular disease. Science, 1949. 109(2835): p. 443.
28. Beet, E.A., Sickle cell disease in the Balovale District of Northern Rhodesia. East Afr Med J, 1946. 23: p. 75-86.
29. Beet, E.A., Sickle cell disease in Northern Rhodesia. East Afr Med J, 1947. 24(6): p. 212-22.
30. Allison, A.C., Protection afforded by sickle-cell trait against subtertian malareal infection. Br Med J, 1954. 1(4857): p. 290-4.
31. Allison, A.C., Malaria in carriers of the sickle-cell trait and in newborn children. Exp Parasitol, 1957. 6(4): p. 418-47.
32. Motulsky, A.G., Hereditary Red Cell Traits and Malaria. Am J Trop Med Hyg, 1964. 13: p. SUPPL147-58.
33. Rucknagel, D.L., K.A. Kalinyak, and M.J. Gelfand, Rib infarcts and acute chest syndrome in sickle cell diseases. Lancet, 1991. 337(8745): p. 831-3.
34. Piel, F.B., et al., Global distribution of the sickle cell gene and geographical confirmation of the malaria hypothesis. Nat Commun, 2010. 1: p. 104.
35. Luzzatto, L., E.S. Nwachuku-Jarrett, and S. Reddy, Increased sickling of parasitised erythrocytes as mechanism of resistance against malaria in the sickle-cell trait. Lancet, 1970. 1(7642): p. 319-21.
36. Miller, M.J., J.V. Neel, and F.B. Livingstone, Distribution of parasites in the red cells of sickle-cell trait carriers infected with Plasmodium falciparum. Trans R Soc Trop Med Hyg, 1956. 50(3): p. 294-6.
37. Friedman, M.J., et al., Plasmodium falciparum: physiological interactions with the human sickle cell. Exp Parasitol, 1979. 47(1): p. 73-80.
38. Kaul, D.K., D. Chen, and J. Zhan, Adhesion of sickle cells to vascular endothelium is critically dependent on changes in density and shape of the cells. Blood, 1994. 83(10): p. 3006-17.
39. Hunt, J.A. and V.M. Ingram, Abnormal human haemoglobins. II. The chymotryptic digestion of the trypsin-resistant core of haemoglobins A and S. Biochim Biophys Acta, 1958. 28(3): p. 546-9.
40. Sanger, F. and A.R. Coulson, A rapid method for determining sequences in DNA by primed synthesis with DNA polymerase. J Mol Biol, 1975. 94(3): p. 441-8.
41. Wollstein, M.K., K.V., Sickle cell anemia. American Journal of Diseases of Childhood, 1928. 36: p. 998-1011.
42. John, A.B., et al., Prevention of pneumococcal infection in children with homozygous sickle cell disease. Br Med J (Clin Res Ed), 1984. 288(6430): p. 1567-70.
43. Lee, A., et al., Improved survival in homozygous sickle cell disease: lessons from a cohort study. BMJ, 1995. 311(7020): p. 1600-2.
44. Johnson, F.L., et al., Bone-marrow transplantation in a patient with sickle-cell anemia. N Engl J Med, 1984. 311(12): p. 780-3.
45. Sullivan, K.M., Walters, M.C., Patience, M., Leisenring, W., Buchanan, G.R., Castro, O., Davies, S.C., Dickerhoff, R., Eckman, J.R., Graham, M.L., Ohene-Frempong, K., Powars, D., Rogers, Z.R., Scott, J.P., Styles, L., Vichinsky, E., Wall, D.A., Wayne, A.S., Wiley, J., Wingard, J., Bunin, N., Camitta, B., Darbyshire, P.J., Dindorf, P., Klingebiel, T., McMahon, L., Mentzer, W.C., Olivieri, N.F., Orringer, E., Parkman, R., Roberts, I.A.G., Sanders, J.E., Smith, F.O. & Storb, R., Collaborative study of marrow transplantation for sickle cell disease: aspects specific for transplantation of hemoglobin disorders. Bone Marrow Transplantation, 1997. 19: p. 102–105.
46. Hill-Kayser, C.E., et al., TBI during BM and SCT: review of the past, discussion of the present and consideration of future directions. Bone Marrow Transplant, 2010. 46(4): p. 475-84.
47. Carr, B.I., et al., Carcinogenicity and haemoglobin synthesis induction by cytidine analogues. Br J Cancer, 1988. 57(4): p. 395-402.
48. Letvin, N.L., et al., Augmentation of fetal-hemoglobin production in anemic monkeys by hydroxyurea. N Engl J Med, 1984. 310(14): p. 869-73.
49. Platt, O.S., et al., Hydroxyurea enhances fetal hemoglobin production in sickle cell anemia. J Clin Invest, 1984. 74(2): p. 652-6.
50. Charache, S., et al., Hydroxyurea: effects on hemoglobin F production in patients with sickle cell anemia. Blood, 1992. 79(10): p. 2555-65.
51. Charache, S., et al., Effect of hydroxyurea on the frequency of painful crises in sickle cell anemia. Investigators of the Multicenter Study of Hydroxyurea in Sickle Cell Anemia. N Engl J Med, 1995. 332(20): p. 1317-22.
52. Elford, H.L., Effect of hydroxyurea on ribonucleotide reductase. Biochem Biophys Res Commun, 1968. 33(1): p. 129-35.
53. Snyder, R.D., The role of deoxynucleoside triphosphate pools in the inhibition of DNA-excision repair and replication in human cells by hydroxyurea. Mutat Res, 1984. 131(3-4): p. 163-72.
54. Wraith, J.E., Ornithine carbamoyltransferase deficiency. Arch Dis Child, 2001. 84(1): p. 84-88.